



Infantile spasms: A frontline guide for pediatricians
Delaying diagnosis and treatment for this early childhood disorder can lead to worse outcomes, so early recognition is crucial.


Newborn baby| Image Credit: © Gary - © Gary - stock.adobe.com.

“[He] was a remarkably fine, healthy child when he was born, and continued to thrive till he was 4 months old. It was at this time that I first observed slight bobbings of the head forward…these bobbings increased in frequency, and at length became so frequent and powerful, as to cause a complete heaving of the head forward to the knees, and then immediately relaxing into the upright position.”1
British physician William James West wrote the above in an 1841 letter to the editor of The Lancet, begging for the medical profession’s attention to a condition afflicting his infant son James.1 This letter was the first published description of what became known as West syndrome, or infantile spasms. The clinical features described—repeated attacks of “bowings and relaxings” increasing in severity and accompanied by developmental regression—are still deeply familiar to any parent or physician caring for a child with infantile spasms.
The primary care pediatrician is the front line for early recognition and treatment initiation in these children. This guide is designed to equip pediatricians with focus on the clinical features, workup, and management of this challenging disorder.
Diagnostic criteria and clinical importance
West syndrome is historically defined as an epilepsy of infancy and early childhood characterized by a triad of (1) pathognomonic seizures (infantile spasms), (2) hypsarrhythmia on an electroencephalogram (EEG), and (3) associated neurodevelopmental arrest or regression. The term “infantile spasms” has been used inconsistently in the literature, to encompass the syndrome or to refer to the seizures alone.2 Given inconsistent terminology as well as variability in clinical and encephalographic presentations, the International League Against Epilepsy proposed a new consensus term for the syndrome: infantile epileptic spasms syndrome (IESS), an epileptic encephalopathy defined by the onset of infantile spasms in infants aged between 1 and 24 months, with peak onset between 3 and 12 months.3 Spasm onset is associated with developmental plateauing or regression. EEG is generally abnormal, often but not always with the chaotic rhythm known as hypsarrhythmia.
The estimated incidence for IESS is 1 in 2400 to 5500 live births.4 Evidence suggests that provider comfort in identifying and managing IESS is lacking, with direct consequences for affected infants and families. In a cross-sectional study of 100 parents of children with infantile spasms, the median time from spasm onset to first visit with any health care provider was 5 days, but the median time from onset to first visit with an “effective provider” (one who provided both accurate diagnosis and prescription for appropriate first-line treatment) was 24.5 days, a delay attributed at least in part to poor awareness of the condition among providers.5 Given that worse outcomes may be associated with even a 1-week delay in treatment from onset, it is critical that pediatric health care providers are proficient in recognizing this condition.6,7
Clinical features
Infantile spasms



An infantile spasm is brief and abrupt, generally 1 to 3 seconds, with muscle contraction that can include the head, neck, trunk, and/or extremities. Spasms can occur in clusters, repeating every 5 to 10 seconds over minutes but, especially early in the disease course, may be isolated and infrequent. They may also be subtle, ranging from a brief head nod or facial/eye movement to a jackknifing of the torso at the waist with contraction of the trunk and extremities. When clustering, they often follow a crescendo-decrescendo pattern of intensity in a series of 2 to 100 spasms. See Table 1 for a summary of spasm features.
The context of the spasm episode may provide other helpful clues for recognition. While spasms can occur anytime, they commonly occur soon after the infant awakens. Behavioral changes around the spasm are common. Caregivers may notice that the infant looks scared or surprised during or immediately before an episode. Following the episode, it is common for the infant to cry, scream, or seem upset.
Home video recording as a clinical tool
Given the length of a typical clinic visit, it is unlikely that the infant will have a spasm in the office. This is where home video recordings (eg, via smartphones) can be extremely helpful. Home video recording, first advised by the Child Neurology Society to streamline IESS management at the onset of the COVID-19 pandemic, has since been endorsed as a continued recommendation toward timely intervention.4 In preparing to evaluate a patient with possible IESS, pediatricians should ask caregivers to record suspected events. Videos should be reviewed prior to the scheduled visit if possible and filed to share with consulting providers including child neurologists. Shortened clips can easily be shared through patient/provider portals in most electronic medical records, whereas longer videos may require secure cloud services.
Age of onset
While the diagnostic criteria for IESS defines an age range of 1 to 24 months, the average age of spasm onset is 6 to 7 months, and most infants will have onset between 3 and 12 months. Although it is possible for IESS to first present after 18 months or before 3 months, it is very uncommon, and spasm-like events should raise concern for another etiology, including conditions other than epilepsy.
Neurodevelopmental changes



Many infants will appear developmentally normal at spasm onset, with neurodevelopmental consequences becoming more apparent as spasms continue. In addition, as risk for IESS is significantly increased in infants with preexisting developmental delay, including some conditions independently associated with developmental delay (eg, tuberous sclerosis complex or trisomy 21), it may be difficult for the caregiver or physician to distinguish initially subtle neurodevelopmental consequences of IESS from antecedent developmental abnormalities.
The differential diagnosis for IESS is broad and includes both neurologic and non-neurologic considerations. Table 2 provides some common mimics for spasms with clinical clues. Given the possible consequences of untreated IESS and evidence for frequent misdiagnosis, it is important to maintain high clinical suspicion, especially in the setting of suspicious features.5,7 Red flag features are highlighted in Table 3.
Workup
Urgency
Clinical suspicion for IESS requires urgent action. Next steps in workup and management should begin within 24 hours, starting with an EEG. Ideally, the pediatrician should immediately reach out to a child neurologist for rapid assessment and EEG. This is best done by phoning the on-call neurologist line at a regional children’s hospital. It is extremely helpful to provide a video clip during this communication. If concerned, the child neurologist will coordinate a same-day EEG, either outpatient or through the emergency department, followed by possible admission.
The national shortage of child neurologists and uneven distribution across the country can make this option difficult. If it is not possible to connect directly with a child neurologist for urgent assessment, other options include directly sending the infant to the emergency department (ED) of a regional children’s hospital with child neurologists on staff for EEG and possible admission (preferred) or sending them to the ED of a local hospital with adult neurologists on staff for the same. If a pediatrician can order a same-day EEG with access to rapid results, that can also help guide the urgency of a neurological consultation.
EEG
The interictal EEG finding most commonly associated with IESS is hypsarrhythmia, which describes a chaotic, disorganized, and high-amplitude background rhythm with asynchronous slowing and multifocal epileptiform discharges. Of note, classic hypsarrhythmia is not always seen nor a requirement for IESS diagnosis. Some infants may show 1 of several variations known together as modified hypsarrhythmia, and others may show epileptiform discharges without altered background. The EEG pattern during the spasm itself is a high-amplitude sharp or slow wave followed by a relative electrodecrement.
Because the interictal EEG is generally abnormal, it is not necessary to capture a spasm during the EEG recording to support the diagnosis. The ideal EEG recording captures a full sleep-wake cycle and so is generally a few hours long. This is because the interictal abnormalities associated with IESS are potentiated during non–rapid eye movement sleep.8 However, EEG abnormalities are often still detectable in the absence of sleep, so shorter EEGs (eg, routine 30-minute) can sometimes be sufficient.
Other elements in workup
Once diagnosis confirmed, the next step in workup is to evaluate for an underlying etiology. Diagnostic workup and treatment initiation are often conducted inpatient, which allows for rapid coordination of the various steps. However, this is not always necessary, especially in cases of clear IESS diagnosis (ie, clinically evident spasms and classic hypsarrhythmia on EEG) with an obvious underlying etiology (ie, previously established trisomy 21 diagnosis).
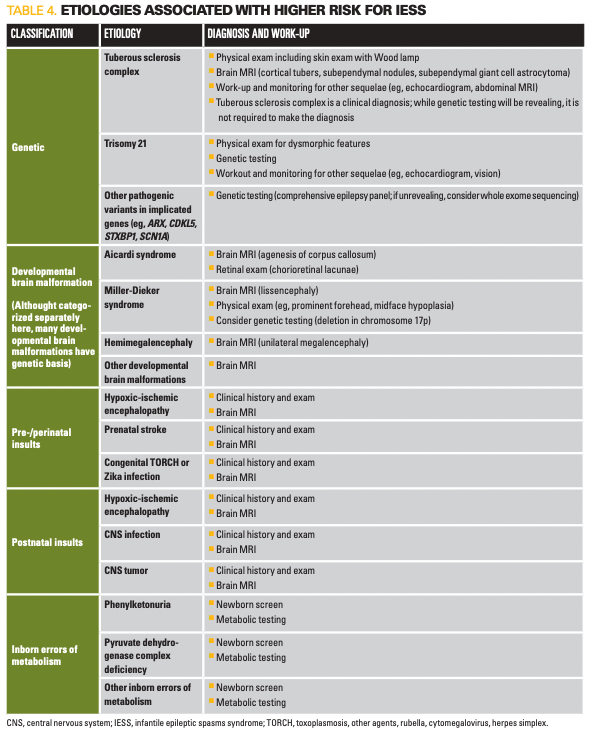
A list of common underlying etiologies is presented in Table 4. Workup for an underlying etiology if not known is important not only because some are associated with other health concerns requiring monitoring and intervention, but also because it can guide management, as some etiologies may respond better to different treatment approaches.9 Identification may also guide appropriate counseling of families, including prognostication and possible genetic counseling.
A detailed clinical history and physical exam should be conducted in all patients. MRI is strongly recommended if not previously obtained, even in cases where etiology appears evident (eg, a child with trisomy 21). This initial workup will identify an underlying etiology in over 50% of presenting infants.10,11 Genetic testing followed by potential metabolic testing will be informative in most of the remaining cases.11 Despite thorough diagnostic evaluation, in a sizable minority of cases a clear etiology will not be identified; with a growing list of genes implicated in IESS, it is likely that many of these are genetic in origin.12
Management
Though etiology can help guide management, it is not necessary to wait for MRI or genetic/metabolic evaluations to initiate therapeutic intervention. The 3 recommended first-line medications for IESS are oral corticosteroids (OCS), adrenocorticotropic hormone (ACTH), and vigabatrin.10,13 In the United States, either oral corticosteroids (typically high-dose prednisolone) or ACTH are generally preferred as initial therapies over vigabatrin in part because of the latter’s adverse effect profile, though there have historically been few trials evaluating their head-to-head efficacy.
A 2021 multicenter study by the National Infantile Spasms Consortium responded to this need by prospectively following 423 children aged 2 to 24 months with new-onset infantile spasms to compare treatment response among ACTH, OCS, vigabatrin, and nonstandard therapy.14 The study concluded that initial treatment with ACTH or OCS is superior to nonstandard therapy. Vigabatrin efficacy was estimated to be between that for ACTH/OCS and nonstandard therapy, but the study was underpowered to evaluate whether vigabatrin was statistically inferior. As previously reported and detailed below, vigabatrin was the superior initial therapy in the setting of tuberous sclerosis complex (TSC).
Two weeks after initiation of first-line therapy, infants should undergo an exam including possible EEG to evaluate for electroclinical treatment response. This is generally done in an outpatient child neurology clinic. Clinical and EEG improvement will guide continued management, including possible dosage modifications, switching of therapies, and combining of therapies. In the following subsections, we will review major therapies with focus on what the primary care pediatrician needs to know.
Steroids (hormonal therapy)



ACTH and OCS were initially used empirically and their mechanisms of action in IESS remain somewhat unclear. ACTH is given via intramuscular injection with dosing based on body surface area. Dosing protocols vary and there is evidence that low to moderate doses (eg, 120 units/m2) may be as effective as high-dose protocols (eg, 40 units/m2), with improved adverse effects.15,16 Standard protocols include daily injections at static dose over 2 to 3 weeks followed by a several-week taper. OCS are given orally and can include prednisolone, prednisone, dexamethasone, and others; in the United States, prednisolone (15 mg/5 mL solution) is generally preferred. For OCS, there is evidence that high-dose therapy (4 to 8 mg/kg/day) is superior to low-dose therapy (1 to 2 mg/kg/day). Similar to ACTH, treatment is generally given for 2 weeks, followed by a gradual taper over 2 to 4 weeks. Dose may be increased if spasms continue after 1 week. In the United Kingdom Infantile Spasms Study (UKISS), infants with continued spasms after 1 week of 40 mg/day prednisolone had the dose increased to 60 mg/day prednisolone for week 2.17 For most infants, we suggest an initial dose of 45 mg/day prednisolone divided 3 times a day (15 mg/dose), with an increase to 60 mg per day prednisolone divided 4 times a day (15 mg/dose) if ineffective after 1 week.18


Both ACTH and high-dose prednisolone have significant adverse effects, including immunosuppression, hypertension, hyperglycemia, stress ulcers and other gastrointestinal irritation, behavioral irritability, disturbed sleep, increased appetite, and adrenal insufficiency. Precautions are taken prior to, during, and following treatment toward these risks, including glucose and blood pressure monitoring and stress ulcer prophylaxis. Following hospital discharge, this will largely fall to the primary care pediatrician, who should aim to see the patient regularly. Patients must also be followed closely for spasm recurrence; even with initially effective treatment response, recurrence rate with steroids is about 33%. Along with an outpatient child neurologist, the pediatrician is a critical ally in this monitoring.
Vigabatrin
Vigabatrin is an irreversible inhibitor of GABA-transaminase that acts by increasing GABA concentration in the central nervous system. Starting dosage is 50 mg/kg/day divided twice daily, which can be slowly uptitrated to a maximum dosage of 150 mg/kg/day divided twice daily. Treatment generally continues for 6 months but in the absence of clinical and/or EEG improvement at the 2-week mark, it may be discontinued in favor of another therapy. Vigabatrin is associated with considerable adverse effects, the most serious being permanent peripheral visual field loss secondary to retinal toxicity. Children on vigabatrin must receive a baseline eye exam within 4 weeks of treatment initiation and subsequent exams every 3 months through treatment and 3 to 6 months following treatment. This exam should be conducted by a pediatric ophthalmologist and should include visual field testing whenever possible. Caregivers should be counseled on other adverse effects including sedation, behavior changes, sleep changes, and weight gain. Vigabatrin is also associated with MRI changes in the brain, which have unclear clinical significance but are generally reversible after conclusion of treatment.19
Vigabatrin is particularly effective in infants with TSC, where it has been found to be superior to ACTH or OCS, and should be the first-choice therapy in these patients. Up to 95% of children with TSC-related IESS will have a complete treatment response.20
Other therapies
Several antiseizure medications have been trialed in IESS, including topiramate, zonisamide, phenobarbital, and levetiracetam. These medications are inferior to first-line therapies and should not typically be used as initial therapies.14 In some cases, children with IESS may have other types of seizures at presentation or will evolve to other seizure types or disorders, precipitating a need for additional medications. Many children with IESS will go on to develop a subsequent seizure disorder. Antiseizure medications in these children should be managed by a child neurologist.
The ketogenic diet is a high-fat, low-carbohydrate diet extensively used in the management of refractory epilepsy, either as a monotherapy or in conjunction with medications. There is considerable evidence that the ketogenic diet can be effective in IESS, and its availability as a liquid formula makes it a viable option for infants. Most of this work has been done in patients with IESS refractory to standard first-line treatment.21,22 In a systematic review of ketogenic diet therapy for IESS, a median of about two-thirds of patients had an over 50% reduction in spasms, and a median of one-third were seizure free.21
Follow-up and prognosis
Unfortunately, even in some cases with spasm resolution, long-term prognosis is often poor. Outcomes include subsequent epilepsy and neurodevelopmental impairment. Etiology may be the most important predictor of outcome.23 Lead time to treatment is another critical and modifiable prognostic factor.23 Several studies to date have found improved developmental/intellectual outcomes with shorter delays between spasm onset and treatment, highlighting the importance of early intervention.6,7,24 In the UKISS study, this relationship was suggested to be dose dependent, where longer lead time durations were associated with a stepwise decline in developmental assessment scores at aged 4 years.6
Children with history of IESS should be followed closely for developmental and neurological sequelae. Psychomotor development should be monitored until at least kindergarten, even in children who appear to have excellent prognoses. It is best to involve a developmental pediatrician in this care as well as to have a low bar for the recruitment of allied specialists including physical, occupational, and speech therapists. While outcomes can be challenging, close allyship among the family, primary pediatrician, neurologist, and broader care team will go far toward creating an environment of support and resilience.
To read more from the April, 2023, issue of Contemporary Pediatrics®, click here.
References
1. West WJ. On a peculiar form of infantile convulsions. The Lancet. 1841;35(911):724-725. doi:10.1016/S0140-6736(00)40184-4
2. Mytinger JR. Definitions and diagnostic criteria for infantile spasms and West syndrome - historical perspectives and practical considerations. Semin Pediatr Neurol. 2021;38:100893. doi:10.1016/j.spen.2021.100893
3. Zuberi SM, Wirrell E, Yozawitz E, et al. ILAE classification and definition of epilepsy syndromes with onset in neonates and infants: position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia. 2022;63(6):1349-1397. doi:10.1111/epi.17239
4. Grinspan ZM, Mytinger JR, Baumer FM, et al. Management of infantile spasms during the COVID-19 pandemic. J Child Neurol. 2020;35(12):828-834. doi:10.1177/0883073820933739
5. Hussain SA, Lay J, Cheng E, Weng J, Sankar R, Baca CB. Recognition of infantile spasms is often delayed: the ASSIST Study. J Pediatr. 2017;190:215-221.e1. doi:10.1016/j.jpeds.2017.08.009
6. O'Callaghan FJ, Lux AL, Darke K, et al. The effect of lead time to treatment and of age of onset on developmental outcome at 4 years in infantile spasms: evidence from the United Kingdom Infantile Spasms Study. Epilepsia. 2011;52(7):1359-1364. doi:10.1111/j.1528-1167.2011.03127.x
7. Auvin S, Hartman AL, Desnous B, et al. Diagnosis delay in West syndrome: misdiagnosis and consequences. Eur J Pediatr. 2012;171(11):1695-1701. doi:10.1007/s00431-012-1813-6
8. Watanabe K, Negoro T, Aso K, Matsumoto A. Reappraisal of interictal electroencephalograms in infantile spasms. Epilepsia. 1993;34(4):679-685. doi:10.1111/j.1528-1157.1993.tb00446.x
9. Messer R, Knupp KG. Infantile spasms: opportunities to improve care. Semin Neurol. 2020;40(2):236-245. doi:10.1055/s-0040-1705121
10. Pellock JM, Hrachovy R, Shinnar S, et al. Infantile spasms: a U.S. consensus report. Epilepsia. 2010;51(10):2175-2189. doi:10.1111/j.1528-1167.2010.02657.x
11. Wirrell EC, Shellhaas RA, Joshi C, et al. How should children with West syndrome be efficiently and accurately investigated? Results from the National Infantile Spasms Consortium. Epilepsia. 2015;56(4):617-625. doi:10.1111/epi.12951
12. Yuskaitis CJ, Ruzhnikov MRZ, Howell KB, et al. Infantile spasms of unknown cause: predictors of outcome and genotype-phenotype correlation. Pediatr Neurol. 2018;87:48-56. doi:10.1016/j.pediatrneurol.2018.04.012
13. Wilmshurst JM, Gaillard WD, Vinayan KP, et al. Summary of recommendations for the management of infantile seizures: task force report for the ILAE Commission of Pediatrics. Epilepsia. 2015;56(8):1185-1197. doi:10.1111/epi.13057
14. Grinspan ZM, Knupp KG, Patel AD, et al. Comparative effectiveness of initial treatment for infantile spasms in a contemporary US cohort. Neurology. 2021;97(12):e1217-e1228. doi:10.1212/WNL.0000000000012511
15. Fayyazi A, Eslamian R, Khajeh A, Dehghani M. Comparison of the effect of high and low doses of adrenocorticotropic hormone (ACTH) in the management of infantile spasms. Iran J Child Neurol. 2020;14(2):17-25.
16. Riikonen R, Lahdetie J, Kokki H. ACTH treatment of infantile spasms: low-moderate- versus high-dose, natural versus synthetic ACTH-a retrospective cohort study. Pediatr Neurol. 2020;111:46-50. doi:10.1016/j.pediatrneurol.2020.06.010
17. Lux AL, Edwards SW, Hancock E, et al. The United Kingdom Infantile Spasms Study comparing vigabatrin with prednisolone or tetracosactide at 14 days: a multicentre, randomised controlled trial. Lancet. 2004;364(9447):1773-1778. doi:10.1016/S0140-6736(04)17400-X
18. Kossoff EH, Hartman AL, Rubenstein JE, Vining EP. High-dose oral prednisolone for infantile spasms: an effective and less expensive alternative to ACTH. Epilepsy Behav. 2009;14(4):674-676. doi:10.1016/j.yebeh.2009.01.023
19. Dracopoulos A, Widjaja E, Raybaud C, Westall CA, Snead OC 3rd. Vigabatrin-associated reversible MRI signal changes in patients with infantile spasms. Epilepsia. 2010;51(7):1297-1304. doi:10.1111/j.1528-1167.2010.02564.x
20. Hancock E, Osborne JP. Vigabatrin in the treatment of infantile spasms in tuberous sclerosis: literature review. J Child Neurol. 1999;14(2):71-74. doi:10.1177/088307389901400201
21. Prezioso G, Carlone G, Zaccara G, Verrotti A. Efficacy of ketogenic diet for infantile spasms: a systematic review. Acta Neurol Scand. 2018;137(1):4-11. doi:10.1111/ane.12830
22. Kossoff EH, Pyzik PL, McGrogan JR, Vining EP, Freeman JM. Efficacy of the ketogenic diet for infantile spasms. Pediatrics. 2002;109(5):780-783. doi:10.1542/peds.109.5.780
23. Riikonen R. Infantile spasms: outcome in clinical studies. Pediatr Neurol. 2020;108:54-64. doi:10.1016/j.pediatrneurol.2020.01.015
24. Kivity S, Lerman P, Ariel R, Danziger Y, Mimouni M, Shinnar S. Long-term cognitive outcomes of a cohort of children with cryptogenic infantile spasms treated with high-dose adrenocorticotropic hormone. Epilepsia. 2004;45(3):255-262. doi:10.1111/j.0013-9580.2004.30503.x




















