



Osteogenesis Imperfecta in a 3-Year-Old Boy
A 3-year-old boy who presents with blue sclerae and a history of tibial fracture following a minor trau- ma (jump from a height of less than 18 inches). Has a long-standing complaint of back pain. Mother remarks that the boy bruises easily. Medical history otherwise unremarkable.


HISTORY
A 3-year-old boy who presents with blue sclerae and a history of tibial fracture following a minor trauma (jump from a height of less than 18 inches). Has a long-standing complaint of back pain. Mother remarks that the boy bruises easily. Medical history otherwise unremarkable.
Not currently taking any medications or vitamins. Was breast-fed for 2 months and was then switched to a regular vitamin D-fortified infant formula. Has not received calcium or vitamin D supplements since starting regular cow's milk and solid food. Currently has a good intake of dairy products.
Patient's mother had multiple fractures throughout her childhood and adolescence. No recent fractures. Mother has had significant back pain since her first pregnancy; also has mild hearing loss and blue sclerae. An 11-month-old healthy sister also noted to have blue sclerae.
PHYSICAL EXAMINATION
Child appears well. Height, just below the 50th percentile; weight, close to the 75th percentile. No dysmorphic features. Results of cardiovascular and abdominal examinations, normal. No skeletal deformity. Hyperflexible joints. Extensive hematomas of varying ages over his shins.
The hereditary disorder osteogenesis imperfecta-commonly known as brittle bone disease-is characterized by excessive fragility of bones and weakness of tissues rich in type I collagen.1 Other major clinical features include skeletal deformity, short stature, dentinogenesis imperfecta, blue sclerae, laxity of ligaments, and hearing loss.2 There are 7 different types of osteogenesis imperfecta: the severity of the clinical presentation varies to some degree among these types.
"WHAT'S YOUR DIAGNOSIS?"
ANSWER: OSTEOGENESIS IMPERFECTA
EPIDEMIOLOGY
Osteogenesis imperfecta occurs in approximately 1 in 20,000 births without racial or ethnic preference.3 Both sexes are equally affected.
GENETICS
The majority of cases have an autosomal dominant mode of inheritance.4,5 Some cases of type II osteogenesis imperfecta with an apparent autosomal recessive mode of inheritance are caused by parental mosaicism and are actually autosomal dominant.4,6 Type VII has an autosomal recessive mode of inheritance.7
PATHOGENESIS
Type I collagen is a heterotrimer composed of 2 a1 chains and a single a2 chain twisted into a tight helical configuration.5 Both chains are approximately 1014 amino acids long and consist of repeats of the amino acid triplet GXY, where G is glycine, X is often proline, and Y is often hydroxyproline.5,7,8 The presence of glycine at every third residue is crucial to the formation of the helix.7 Type I collagen is the major structural protein of the extracellular matrix of bone, skin, and tendons.9
Most cases of osteogenesis imperfecta result from mutations in one or the other of the 2 genes (COL1A1 and COL1A2), which encode the a1 and a2 chains of type I procollagen.4 The COL1A1 gene is located on the long arm of chromosome 17, and the COL1A2 gene on the long arm of chromosome 7.10 Such mutations may alter the amino acid sequence of either the triple helical domain or the carboxyl-terminal propeptide of the encoded pro a chain,8 resulting in decreased synthesis of structurally normal collagen or structurally abnormal collagen.10,11 Diseases associated with decreased synthesis of structurally normal collagen are usually milder than those associated with synthesis of structurally abnormal collagen.10,11
CLINICAL CLASSIFICATION AND MANIFESTATIONS
The current classification divides osteogenesis imperfecta into 7 types based on clinical and radiographic criteria.2,7,12



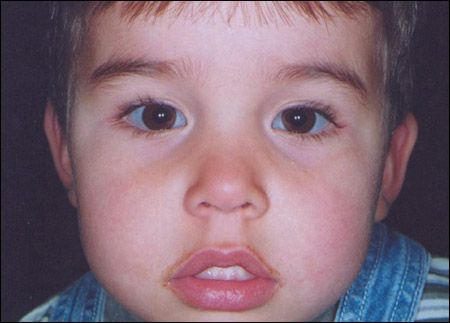
Figure 1 – The child in our case history had osteogenesis imperfecta type IB. The child's mother also had mild hearing loss and blue sclerae, and an 11-monthold healthy sister had blue sclerae.
Type I is the most common variant-and the mildest.12,13 It is characterized by generalized mild to moderate osteoporosis and bone fragility, distinctly blue sclerae throughout life, and pre-senile hearing loss.4,12,14 Type I is further divided into 2 subgroups depending on the absence (IA) or presence (IB) of dentinogenesis imperfecta.4,14 Affected patients may have mild short stature, ligamentous laxity, easy bruising, delayed closure of fontanelles, and an increased incidence of hernia.15 Life expectancy and fertility are unaffected.16 The child in the case we present here had osteogenesis imperfecta type IB (Figure 1).
Type II is the perinatal lethal form. Approximately 50% of affected infants are born prematurely or are stillborn.17 The remaining infants usually die of respiratory insufficiency, congestive heart failure, and/orinfection during the first year of life.16,17 Type II is characterized by extreme bone fragility. Multiple intrauterine fractures of long bones are common.15 Affected infants have a triangular face, dark blue-gray sclerae, beaked nose, soft calvarium, wormian bones in the skull, micromelia, bowed legs, small thorax, and beaded ribs.15-17


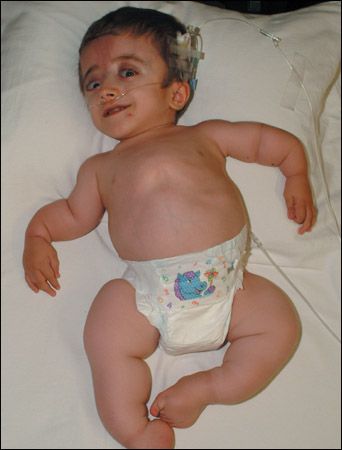
Figure 2 – The most severe nonlethal form of osteogenesis imperfecta, type III disease is characterized by severe bone fragility; multiple fractures; progressive marked deformity of the spine and long bones; and severe short stature associated with vertebral compression, kyphosis, deformity of limbs, and disrupted growth.
Type III, the most severe nonlethal form, is characterized by severe bone fragility, multiple fractures, and progressive marked deformity of the spine and long bones (Figure 2).3 Severe short stature may result from vertebral compression, kyphosis, deformity of limbs, and disrupted growth.4,16 Other characteristic findings include a triangular face, relative macrocephaly, wormian bones, large fontanelles, frontal bossing, scoliosis, flaring of the rib cage at the base, dentinogenesis imperfecta, and blue sclerae.4,15 Blue sclerae tend to become less blue with age. Pre-senile hearing loss is less common than in type I disease. Most children are wheelchair-dependent by adolescence.17
In type IV, bone fragility with resultant fractures and short stature tend to be more severe than in type I but less severe than in type III.1 The hallmarks of osteogenesis imperfecta type IV are tibial bowing and normal sclerae.3,12 Like type I, the frequency of fracture decreases at puberty.16,17 Pre-senile hearing loss is less frequent and dentinogenesis imperfecta is more common than in type I.10 Type IV is divided into 2 subgroups according to whether the patient has normal teeth (IVA) or dentinogenesis imperfecta (IVB).3,10
Type V is characterized by osteoporosis, hypertrophic calluses after fractures, early ossification of the interosseous membrane of the forearms and legs, and radiodense metaphyseal bands.4,7,12,15
Type VI is characterized by frequent fractures with resultant deformities of long bones and vertebral compressions, absence of dentinogenesis imperfecta, and normal-colored sclerae.7 Histologically, there is an abundance of osteoid tissue and a fish-scale pattern of lamellation.7
Type VII manifests with clinical and histologic features similar to those of type VI. Rhizomelic limb shortening and coxa vara are characteristic features.7
COMPLICATIONS
Depending on the severity of osteogenesis imperfecta, affected persons are prone to recurrent fractures with resultant deformities of the spine and limbs.4,13,16 Patients with type III and type IV are often wheelchair-dependent by adolescence. Pre-senile hearing loss is frequent. Recurrent pneumonia, cardiac failure, and respiratory insufficiency may develop in those patients with type II disease who have decreased thoracic size.13,15 Neurologic complications include intracranial hemorrhage after vaginal delivery, basilar impression, brain stem compression, and hydrocephalus.10,15 The incidence of cardiac valvularproblems, such as aortic and mitral incompetence, is increased among persons with osteogenesis imperfecta.16
Management of Osteogenesis Imperfecta Treatment is mainly symptomatic and supportive. Physiotherapy, rehabilitation, casting, bracing, splinting, and timely surgical correction of deformities are the mainstays.2,17 The patient in our case history was treated in this manner.Recently, intravenous (pamidronate) or oral (alendronate) bisphosphonates have been shown to increase bone density, reduce the incidence of fractures, diminish chronic pain, and improve mobility in children with osteogenesis imperfecta.2,12,13,26,27 Bisphosphonates are synthetic analogs of pyrophosphate that bind to hydroxyapatite crystals in the bone mineral.7,26 These compounds inhibit osteoclastic activity and prevent bone resorption.13,26The use of mesenchymal stromal cell therapy and gene therapy in the treatment of osteogenesis imperfecta is still experimental.4,5,13
DIAGNOSTIC STUDIES
Prenatal diagnosis of severe osteogenesis imperfecta is possible with ultrasonography as early as 16 weeks of gestation.15 Characteristic findings include multiple fractures, deformed long bones, severely decreased long bone length, decreased echogenicity of the calvarium and skeleton, and thoracic abnormalities.1,10 For a fetus at known risk for osteogenesis imperfecta, chorionic villus cells obtained by sampling with the aid of ultrasonography can be used for biochemical or molecular studies.10,15
Radiographs may show osteopenia/osteoporosis, multiple fractures, bone deformities, or wormian bones in the skull. Wormian bones are frequent but not universal in patients with osteogenesis imperfecta.18 Dual-energy x-ray absorptiometry helps assess bone density.1,16
Serum calcium, phosphorus, and parathyroid hormone levels are usually normal in patients with osteogenesis imperfecta.19 Serum alkaline phosphatase levels may be normal or elevated.15 Hypercalcemia may result from immobilization. Hypercalciuria is common and seems to be related to the severity of the disease and to the effects of prolonged immobilization.16,20
Biochemical and molecular studies on cultured dermal fibroblasts from a skin biopsy may be used to identify the presence and type of osteogenesis imperfecta when the diagnosis is in doubt.10,16 However, routine skin biopsy for diagnosis is unwarranted.21
DIFFERENTIAL DIAGNOSIS
In the neonatal period, the differentiation between osteogenesis imperfecta and congenital hypophosphatasia can be difficult because severe deficiency of ossification, fractures, and shortening of long bones can be seen in both conditions. Patients with congenital hypophosphatasia, however, have very low serum alkaline phosphatase levels and large quantities of phosphoethanolamine in the urine.
Camptomelic dysplasia may mimic osteogenesis imperfecta in that it manifests with bowing of extremities.10 It can be distinguished from osteogenesis imperfecta by hypoplastic or absent scapulae and fibulae, hypoplastic thorax, absent thoracic pedicles, brachydactyly, and micrognathia.10 Fractures are not a feature of camptomelic dysplasia.
Child abuse must be considered in children who have unexplained fractures.22 Clinical features that favor child abuse include retinal hemorrhage, intracranial injury, cigarette burn marks, lacerations, metaphyseal corner fractures, and signs of sexual abuse.1 A family history of osteogenesis imperfecta, as well as the presence of blue sclerae, dentinogenesis imperfecta, osteoporosis, and wormian bones in the skull, suggests osteogenesis imperfecta.10,23 When the diagnosis of osteogenesis imperfecta is in doubt, collagen type I analysis can be very helpful.2 Bear in mind that child abuse and osteogenesis may coexist, however.24,25
References:
REFERENCES:
1. Tosi LL. Osteogenesis imperfecta. Curr Opin Pediatr.1997;9:94-99.
2. Rauch F, Glorieux FH. Osteogenesis imperfecta. Lancet. 2004;363:1377-1385.
3. Engelbert RH, van der Graaf Y, van Empelen R, et al. Osteogenesis imperfecta in childhood: impairment and disability. Pediatrics. 1997;99:e3.
4. Cole WG. Advances in osteogenesis imperfecta. Clin Orthop Relat Res. 2002; 1:6-16.
5. Marini JC, Gerber NL. Osteogenesis imperfecta. Rehabilitation and prospects for gene therapy. JAMA. 1997;277:746-750.
6. Cohn DH, Starman BJ, Blumberg B, Byers PH. Recurrence of lethal osteogenesis imperfecta due to parental mosaicism for a dominant mutation in a human type I collagen gene (COL1A1). Am J Hum Genet. 1990;46:591-601.
7. Zeitlin L, Fassier F, Glorieux FH. Modern approach to children with osteogenesis imperfecta. J Pediatr Orthop B. 2003;12:77-87.
8. Byers PH. Osteogenesis imperfecta: perspectives and opportunities. Curr Opin Pediatr.2000;12:603-609.
9. Devogelaer JP. New uses of bisphosphonates: osteogenesis imperfecta. Curr Opin Pharmacol. 2002;2:748-753.
10. Ablin DS. Osteogenesis imperfecta: a review. Can Assoc Radiol J. 1998;49: 110-123.
11. Antoniazzi F, Bertoldo F, Mottes M, et al. Growth hormone treatment in osteogenesis imperfecta with quantitative defect of type I collagen synthesis. J Pediatr. 1996;129:432-439.
12. Glorieux FH. The use of bisphosphonates in children with osteogenesis imperfecta. J Pediatr Endocrinol Metab.2001;14(suppl 6):1491-1495.
13. Niyibizi C, Smith P, Mi Z, et al. Potential of gene therapy for treating osteogenesis imperfecta. Clin Orthop Relat Res. 2000;1(suppl 379):S126-S133.
14. Engelbert RH, Uiterwaal CS, Gulmans VA, et al. Osteogenesis imperfecta in childhood: prognosis for walking. J Pediatr. 2000;137:397-402.
15. Marini JC. Osteogenesis imperfecta. In: Behrman RE, Kliegman RM, Jenson HB, eds. Nelson Textbook of Pediatrics. Philadelphia: WB Saunders Co; 2004:336-338.
16. Antoniazzi F, Mottes M, Fraschini P, et al. Osteogenesis imperfecta: practical treatment guidelines. Paediatr Drugs. 2000;2:465-488.
17. Bender LH. Osteogenesis imperfecta. Orthop Nurs. 1991;10:23-32.
18. Smith R. Osteogenesis imperfecta, non-accidental injury, and temporary brittle bone disease. Arch Dis Child. 1995;72:169-176.
19. Zaleske DJ. Osteogenesis imperfecta. In: Morrissy RT, Weinstein SL, eds. Lovell and Winter's Pediatric Orthopaedics. Philadelphia: Lippincott Williams & Wilkins; 2001:205-210.
20. Williams CJ, Smith RA, Ball RJ, Wilkinson H. Hypercalcaemia in osteogenesis imperfecta treated with pamidronate. Arch Dis Child. 1997;76:169-170.
21. Steiner RD, Pepin M, Byers PH. Studies of collagen synthesis and structure in the differentiation of child abuse from osteogenesis imperfecta. J Pediatr. 1996;128:542-547.
22.Frasier LD, Herman BE. 5-Year-old with fractured femur and multiple bruises. Consultant For Pediatricians. 2005;4:40-43.
23. Paterson CR, McAllion SJ. Osteogenesis imperfecta in the differential diagnosis of child abuse. Br Med J. 1989;299:1451-1454.
24. Kasim MS, Cheah I, Sameon H. Osteogenesis imperfecta and non-accidental injury: problems in diagnosis and management. Med J Malaysia. 1995;50:170-175.
25. Knight DJ, Bennet GC. Non-accidental injury in osteogenesis imperfecta: a case report. J Pediatr Orthop. 1990;10:542-544.
26. Allgrove J. Bisphosphonates. Arch Dis Child.1997;76:73-75.
27. Chevrel G, Meunier PJ. Osteogenesis imperfecta: lifelong management is imperative and feasible. Joint Bone Spine. 2001;68:125-129.











Recognize & Refer: Hemangiomas in pediatrics
July 17th 2019Contemporary Pediatrics sits down exclusively with Sheila Fallon Friedlander, MD, a professor dermatology and pediatrics, to discuss the one key condition for which she believes community pediatricians should be especially aware-hemangiomas.





