



Kawasaki disease: AHA statement and recommendations
Pediatricians must suspect Kawasaki disease (KD) in children with prolonged unexplained fever. This article reviews the latest scientific statement on KD from the American Heart Association that is of practical importance for all clinicians.


Figure 1
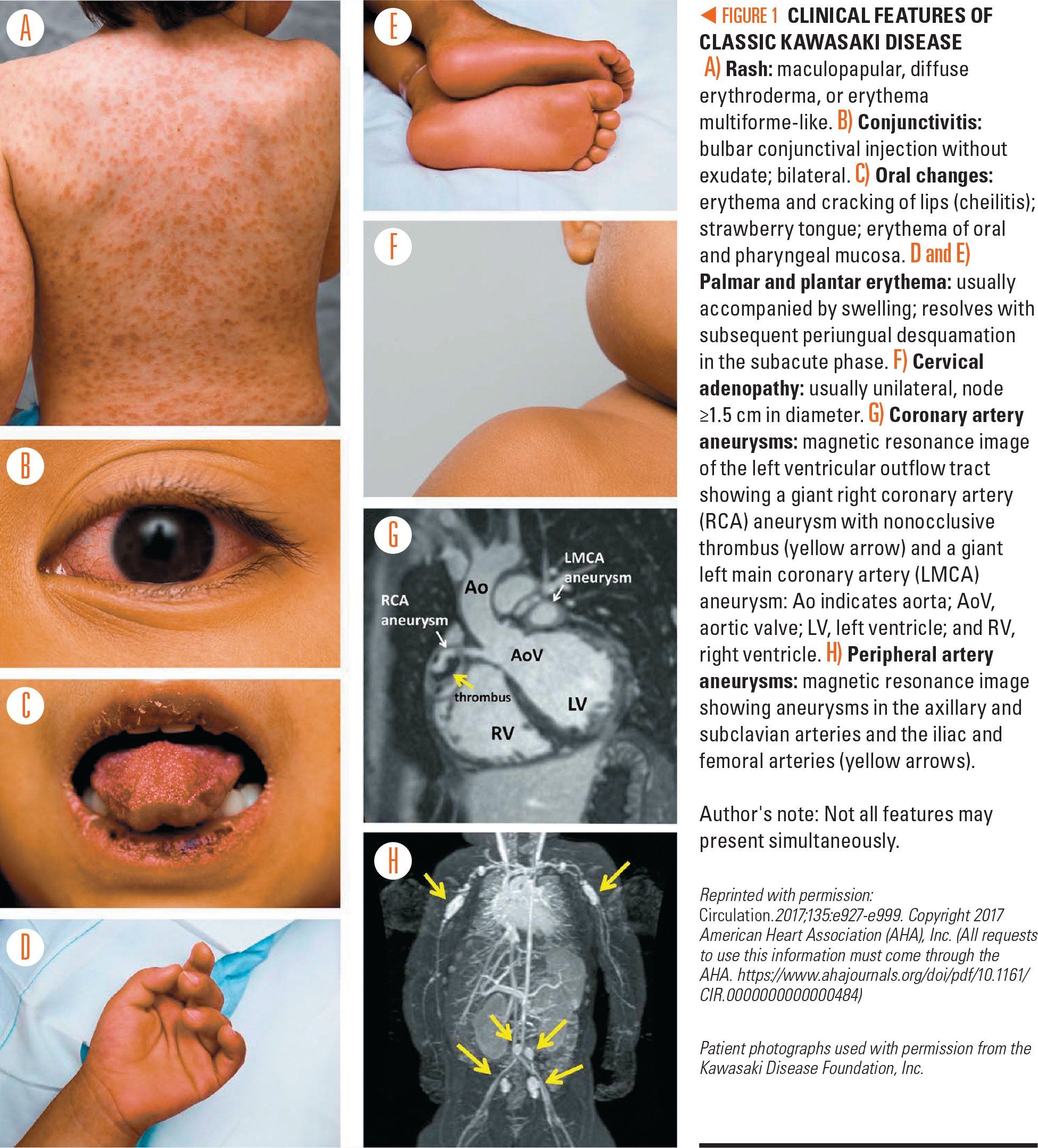


Figure 2
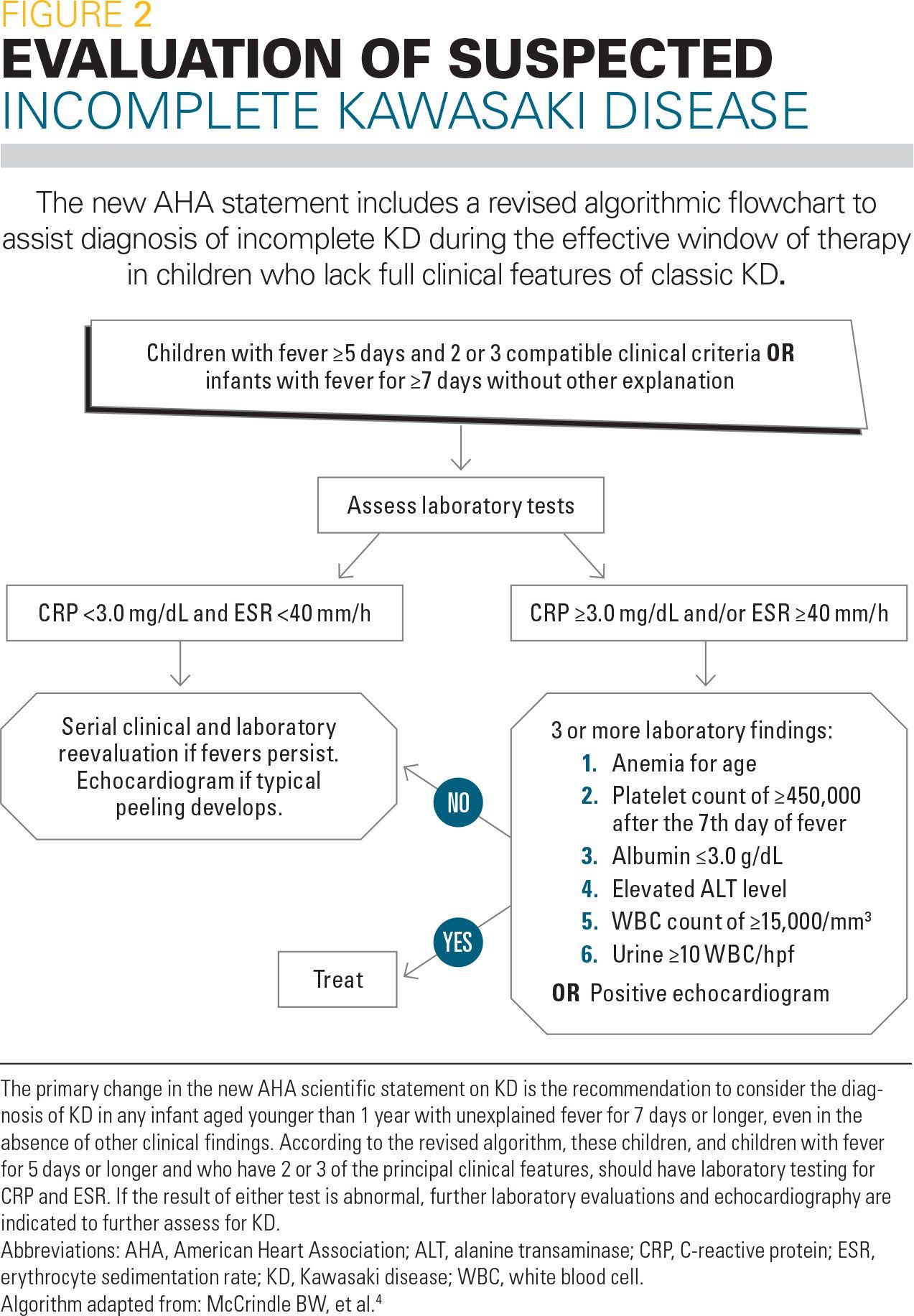


Table 1



Table 2


Kawasaki Disease: A parents' guide video

Kawasaki disease (KD) is an acute systemic vasculitis that occurs predominantly in children aged younger than 5 years. Exact information about the incidence of KD is lacking, but in developed countries it remains the most common cause of acquired heart disease in children.
The incidence of KD varies by race/ethnicity, being highest in Asian children, intermediate in black and Hispanic children, and lowest among white children.1 The disease is estimated to occur in 1 in 1000 US children by age 5 years.2 Therefore, in a large pediatric practice caring for 1000 to 2000 non-Asian children in this age group, 1 to 2 cases would likely be diagnosed over a 5-year period. The attack rate is much higher in Asian children: 1 in 70 Japanese children develop KD by age 5 years.3 Therefore, in parts of the United States with high Asian populations, the index of suspicion for KD should be much higher, and many more cases will be observed.
The etiology of KD remains unknown and there is still no specific test for its diagnosis. Left untreated, KD is associated with the development of potentially fatal coronary artery aneurysms in at least 25% of cases.4 Because early treatment initiation improves the prognosis, it is critical that pediatricians maintain a proper index of suspicion for KD in children with prolonged unexplained fever.
In 2004, the American Academy of Pediatrics and the American Heart Association (AHA) issued a clinical report on KD diagnosis, treatment, and long-term management as guidance for clinicians rendering pediatric care.5 In 2017, the AHA published a new scientific statement reviewing recent evidence and containing updated recommendations for practitioners.4 This article highlights information from the 2017 AHA statement and other recent information that is of practical importance for pediatricians.
Diagnosis
The criteria for diagnosing KD were not changed in the new AHA statement. Classic KD is diagnosed when a child has fever for at least 5 days along with at least 4 of the following 5 principal clinical features (Figure 1):
· Bilateral conjunctival injection.
· Changes of the oral cavity and lips.
· Polymorphous rash.
· Cervical lymphadenopathy.
· Erythema of the extremities.
It is important to keep in mind that any of the principal clinical features could have resolved by the time that the patient presents in the office, and so it is important to take a thorough history.
The new AHA statement includes a revised algorithmic flowchart to assist the diagnosis of incomplete KD during the effective window of therapy in children who lack full clinical features of classic KD (Figure 2).4 The primary change in the new statement is the recommendation to consider the diagnosis of KD in any infant aged younger than 1 year with unexplained fever for 7 days or longer, even in the absence of other clinical findings. According to the revised algorithm, these children, and children with fever for 5 days or longer and who have 2 or 3 of the principal clinical features, should have laboratory testing for C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR). If the result of either test is abnormal, further laboratory evaluations and echocardiography are indicated to further assess for KD.
Pediatricians often may encounter children with fever for 5 days accompanied by rash and red eyes in their practice. Although the vast majority of these children do not have KD, pediatricians should still consider the possibility of KD whenever a patient has prolonged unexplained fever accompanied by any of the principal features of KD. In such patients, initial laboratory assessment such as a complete blood count (CBC) with differential, ESR, and CRP may be useful in determining whether further consideration of a KD diagnosis is warranted.
Regarding the echocardiogram, the new statement is more specific than its predecessor about use of sedation. It recommends repeating the study under sedation as soon as possible within 48 hours after diagnosis and initial treatment if a first echocardiogram was done without sedation and was of poor quality.
The new statement also emphasizes that the echocardiogram report provides a quantitative assessment of luminal dimensions that are normalized as z scores adjusted for body surface area and not merely a qualitative description of vessel appearance. Studies of coronary artery dimensions in febrile children with non-KD illnesses confirm that coronary artery z scores greater than 2.5 are very specific for the diagnosis of KD.6,7
Treatment recommendations
Primary therapy
Research continues to support the need for early diagnosis and treatment of KD, with initiation of intravenous immunoglobulin (IVIG) well before the tenth day of illness. Table 1 summarizes recommendations from the AHA statement pertaining to primary therapy.
There has been long-standing interest in the potential efficacy of using a corticosteroid as an adjunct to IVIG based on recognition that KD results in coronary arterial inflammation. A first randomized trial exploring this approach was conducted in the United States and found no benefit from adding a single high dose of methylprednisolone to IVIG for primary therapy of KD.8 More recently, the Randomized Controlled Trial to Access Immunoglobulin Plus Steroid Efficacy for Kawasaki Disease (RAISE) study, a Japanese multicenter trial enrolling patients at high risk of developing coronary artery abnormalities, demonstrated a significantly lower incidence of coronary artery abnormalities at 1 month after KD onset in the group that received a more prolonged (2- to 3-week) course of adjunctive prednisolone.9
Whereas illness severity scores (eg, the Kobayashi score) can accurately identify high-risk patients in Japanese populations, they have low sensitivity and cannot be reliably implemented in non-Japanese children.10,11 However, certain children with KD, such as infants aged younger than 6 months and those aged 8 years and older, are known to be at particularly high risk of developing coronary artery abnormalities following KD.12 The new AHA statement recommends that single-dose methylprednisolone should not be administered with IVIG as routine primary therapy for patients with KD. It states that the addition of a longer course of corticosteroids (eg, tapering over 2 to 3 weeks) to IVIG and aspirin may be considered for treatment of high-risk patients with acute KD, when such high risk can be identified in patients before initiation of treatment. In the RAISE study, prednisolone was initiated at 2 mg/kg/day and tapered over 2 to 3 weeks depending on fever response, decrease in CRP levels, and clinical course.8
The optimal dose of aspirin for acute KD remains controversial. The new AHA statement continues to recommend administering moderate- (30-50 mg/kg/d) to high-dose (80-100 mg/kg/d) aspirin until the patient is afebrile.4 Some recent research, however, suggests that low-dose aspirin is not inferior to high-dose aspirin for the treatment of acute KD.12,13
A multicenter retrospective study including approximately 1200 children with acute KD seen between 2004 and 2015 and treated with IVIG within 10 days of fever onset found no difference in the risk of coronary artery abnormalities comparing cohorts receiving low-dose (3-5 mg/kg/d) or high-dose (80 mg/kg/d) aspirin.13 A retrospective study including 249 patients treated at 2 centers in Canada found children receiving low-dose aspirin (3-5 mg/kg/d) were 3 times more likely to receive IVIG retreatment compared with children receiving high-dose acetylsalicylic acid (ASA; 80-100 mg/kg/d).14 Both the mean duration of hospital stay and incidence of coronary artery aneurysms, however, were similar in the 2 groups. Further study is needed to provide clarity on this issue.
Rescue therapy
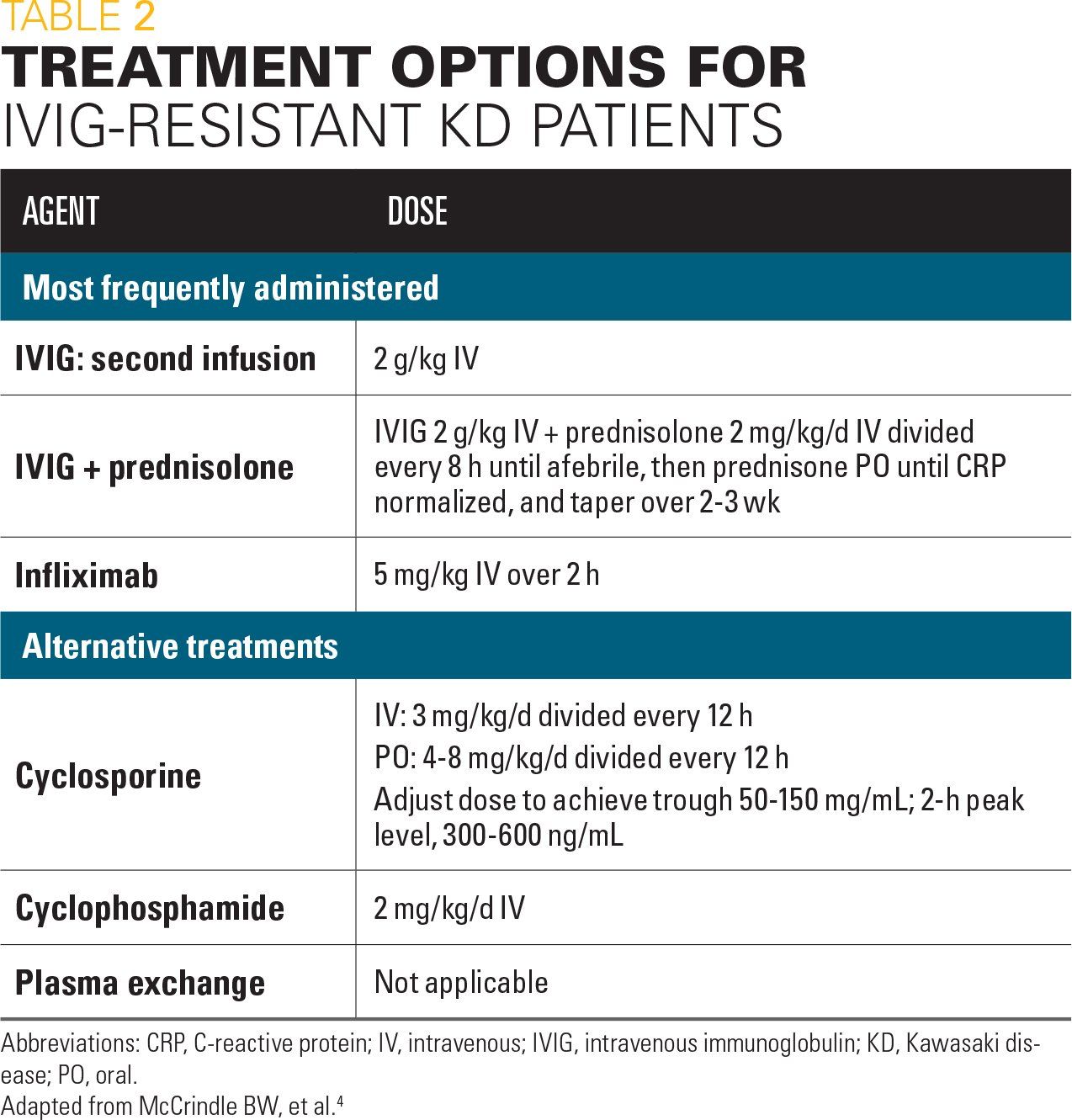
Further study is also needed to develop evidence-based recommendations on rescue therapy for IVIG-resistant patients, defined as those having persistent or recrudescent fever at least 36 hours and less than 7 days after completing the IVIG infusion. The new AHA statement provides a discussion of additional therapy options, including a second dose of IVIG; high-dose pulse steroids; a longer tapering course of prednisolone or prednisone with IVIG and aspirin; infliximab; cyclosporine; other immunomodulatory monoclonal antibody therapy; cytotoxic agents (eg, cyclophosphamide); and (rarely) plasma exchange (Table 2).4 Information about use of these agents is from small studies, retrospective reviews, or case series, and none have been evaluated in any rigorous prospective clinical trials.
Because the clinical symptoms of KD are self-limited, it is difficult to determine whether patients who apparently responded to rescue therapies represent true clinical responses. The small number of IVIG nonresponders and the high cost of performing prospective clinical studies have been obstacles to conducting controlled trials of rescue therapies. A phase 3 multicenter study comparing infliximab to a second dose of IVIG in IVIG-resistant patients is now under way [NCT03065244]. The identification of optimal intervention for children resistant to IVIG would likely be facilitated by elucidation of the etiology and pathogenesis of KD.
Long-term management
Newer information from histopathologic studies of coronary arteries obtained from KD patients provides a better understanding of specific events in damaged arteries and the potential for children with significant acute damage to develop serious complications over time.15 The researchers in this study described a 3-step model of KD arteriopathy culminating with luminal myofibroblastic proliferation that can lead to progressive arterial stenosis, thrombus formation, and myocardial infarction. This process reinforces the importance of ongoing periodic cardiology surveillance for all KD children with an aneurysm and highlights the potential pitfall of incorrectly interpreting normalization of the luminal dimension on an echocardiogram as being equivalent to return to a normally functioning artery.
The new scientific statement also includes a section on transition to adult care.2 As with other pediatric heart diseases, it is important to provide the patient who develops coronary artery aneurysms with a well-developed plan for transitioning to adult cardiology care.
The future
Research investigating genetic associations for KD susceptibility and treatment response are corroborating previous results and generating some interesting new findings. These studies have identified single nucleotide polymorphisms of several genes involved in immune response, including ITPKC, CASP3, FCGR2A, HLA, BLK, CD40, and, most recently, ORA1.16 It is likely that additional susceptibility genes will be identified in future studies.
In addition, evidence associating risk of aneurysm formation with variants in genes involved in the transforming growth factor-β pathway has provided a rationale for investigating the potential for statin treatment to decrease coronary artery inflammation and mitigate arterial damage.4 So far, understanding of genetic associations with KD has no impact for patient care.
Identification of the etiologic agent(s) remains the holy grail for achieving future advances in KD diagnosis, treatment, and prevention. New data continue to support the theory that KD is caused by a novel virus without substantial homology to known viruses.17 Developments in molecular technologies should facilitate attainment of this elusive goal that holds the key to reducing the burdens of KD.
References:
1. Uehara R, Belay Ed. Epidemiology of Kawasaki disease in Asia, Europe, and the United States. J Epidemiol. 2012;22(2):79-85.
2. Holman RC, Belay ED, Christensen KY, Folkema AM, Steiner CA, Schonberger LB. Hospitalizations for Kawasaki syndrome among children in the United States, 1997-2007. Pediatr Infect Dis J. 2010;29(6):483-488.
3. Makino N, Nakamura Y, Yashiro M, et al. Epidemiological observations of Kawasaki disease in Japan, 2013-2014. Pediatr Int. March 2, 2018. Epub ahead of print.
4. McCrindle BW, Rowley AH, Newburger JW, et al.; American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee of the Council on Cardiovascular Disease in the Young; Council on Cardiovascular and Stroke Nursing; Council on Cardiovascular Surgery and Anesthesia; and Council on Epidemiology and Prevention. Diagnosis, treatment, and long-term management of Kawasaki disease: a scientific statement for health professionals from the American Heart Association. Circulation. 2017;135(17):e927-e999.
5. Newburger JW, Takahashi M, Gerber MA, et al.; Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Pediatrics. 2004;114(6):1708-1733. Erratum in: Pediatrics. 2005;115(4):1118.
6. Muniz JC, Dummer K, Gauvreau K, Colan SD, Fulton DR, Newburger JW. Coronary artery dimensions in febrile children without Kawasaki disease. Circ Cardiovasc Imaging. 2013;6(2):239-244.
7. Bratincsak A, Reddy VD, Purohit PJ, et al. Coronary artery dilation in acute Kawasaki disease and acute illnesses associated with fever. Pediatr Infect Dis J. 2012;31(9):924-926.
8. Newburger JW, Sleeper LA, McCrindle BW, et al; Pediatric Heart Network Investigators. Randomized trial of pulsed corticosteroid therapy for primary treatment of Kawasaki disease. N Engl J Med. 2007;356(7):663-675.
9. Kobayashi T, Saji T, Otani T, et al; RAISE study group investigators. Efficacy of immunoglobulin plus prednisolone for prevention of coronary artery abnormalities in severe Kawasaki disease (RAISE study): a randomised, open-label, blinded-endpoints trial. Lancet. 2012;379(9826):1613-1620.
10. Sleeper LA, Minich LL, McCrindle BM, et al; Pediatric Heart Network Investigators. Evaluation of Kawasaki disease risk-scoring systems for intravenous immunoglobulin resistance. J Pediatr. 2011;158(5):831.e3-835.e3.
11. Kobayashi T, Inoue Y, Takeuchi K, et al. Prediction of intravenous immunoglobulin unresponsiveness in patients with Kawasaki disease. Circulation. 2006;113(22):2606-2612.
12. Manlhiot C, Yeung RS, Clarizia NA, Chahal N, McCrindle BW. Kawasaki disease at the extremes of the age spectrum. Pediatrics. 2009;124(3):e410-e415.
13. Dallaire F, Fortier-Morissette Z, Blais S, et al. Aspirin dose and prevention of coronary abnormalities in Kawasaki disease. Pediatrics. 2017;139(6):e20170098.
14. Dhanrajani A, Chan M, Pau S, Ellsworth J, Petty R, Guzman J. Aspirin dose in Kawasaki disease-the ongoing battle. Arthritis Care Res (Hoboken). December 29, 2017. Epub ahead of print.
15. Orenstein JM, Shulman ST, Fox LM, et al. Three linked vasculopathic processes characterize Kawasaki disease: a light and transmission electron microscopic study. PLoS One. 2012;7(6):e38998.
16. Onouchi Y. The genetics of Kawasaki disease. Int J Rheum Dis. 2018;21(1):26-30.
17. Rowley AH, Wylie KM, Kim KY, et al. The transcriptional profile of coronary arteritis in Kawasaki disease. BMC Genomics. 2015;16:1076.
























Pediatric cancer survivors and cardiovascular toxicity, disease risk
March 21st 2025Pediatric cancer survivors can be vulnerable to cardiovascular disease in the short- or long-term with increased recognition of cardiotoxic cancer treatments, as 5-year survival rates for children are greater than 85%.


