



Selective IgA Deficiency in Children: Clinical Manifestations, Evaluation, and Management
Selective IgA deficiency (SIGAD) is the most common immunodeficiency disorder; it affects about 1 in 200 to 900 persons. Most affected children are asymptomatic.
ABSTRACT: Consider an evaluation for IgA deficiency in children older than 6 months who have recurrent bacterial infections in the upper or lower respiratory tract. This evaluation includes a complete blood cell count and measurement of peripheral blood B cells, T cells, NK cells, and levels of serum immunoglobulins. Patients with recurrent infections may benefit from prophylactic antibiotic therapy given for 6 months-or given only during the winter months in those with seasonal patterns of infections. Long-term follow-up to identify other immunological abnormalities, autoimmune disease, and malignancy is required. Antibodies against IgA are known to occur in patients with selective IgA deficiency; such patients are at risk for severe anaphylactic transfusion reactions.
Selective IgA deficiency (SIGAD) is the most common immunodeficiency disorder; it affects about 1 in 200 to 900 persons.1,2 Most affected children are asymptomatic. However, in rare patients, the deficiency leads to an increased incidence of infection, particularly sinopulmonary infection.
About 20% to 25% of patients have a family history of either SIGAD or common variable immunodeficiency (CVID).1 SIGAD, CVID, selective IgG subclass deficiencies, and a syndrome of recurrent sinopulmonary infections with normal serum immunoglobulin all seem to share the same group of gene defects.3 In this article, we review the causes and clinical manifestations of SIGAD and recommendations regarding evaluation and management.
PHYSIOLOGY OF IgA
SIGAD is defined as a serum IgA level of less than 7 mg/dL with normal serum IgG and IgM levels in a patient older than 4 years in whom other causes of hypogammaglobulinemia have been excluded.1 Secretion of IgA. IgA is the predominant antibody class found in bodily secretions, including intestinal and bronchial secretions, saliva, tears, prostatic fluid, and breast milk.4 It is secreted by B cells in submucosal lymphoid tissues, Peyer patches, and tonsils. Secretory IgA binds to and neutralizes intestinal pathogens and their toxic products, including the fimbrial proteins of Escherichia coli and Clostridium difficile toxins.5 Another essential function of intestinal mucosal IgA is the restriction of intestinal bacterial flora to the gut lumen. Normally, IgA antibodies are not found in serum unless bacterial flora invade the bloodstream in a pathological state.5
Role in immune defense. IgA antibodies prevent the attachment of organisms and toxins to epithelial cells.6 The principal function of IgA antibodies is to protect the extracellular spaces of the internal tissues by facilitating phagocytosis in mucosal areas-thereby preventing GI and respiratory tract infections.
CAUSES OF IgA DEFICIENCY
IgA deficiency is a genetic disorder with a heterogeneous clinical presentation. In children with SIGAD, B cells expressing surface IgA are present, but they are developmentally arrested. The clinical heterogeneity may be partially reflected in the extent of an arrest in B cell differentiation and, in some cases, an associated cellular immune defect. The inability of the B cells to differentiate into IgA-secreting plasma cells may be caused by the deficiency of cytokines, such as interleukin (IL)-4, IL-6, IL-7, and IL-10.7
Genetic defects. A mutation in the tumor necrosis factor receptor family member TACI (transmembrane activator and calcium-modulator and cyclophilin ligand interactor) was the first genetic abnormality found in patients with SIGAD. The function of TACI is to facilitate isotype switching in B cells.8 This mutation has also been observed in some patients with CVID, which may explain why some cases of SIGAD can progress to CVID.9 Function-loss mutations in several non–major histocompatibility complex (MHC) genes have been found in families with SIGAD or CVID disorders, in which progression may occur from normal to CVID or to SIGAD with or without IgG subclass deficiency.10 Familial grouping of IgA deficiency has also been found; however, the pattern of inheritance has not yet been clearly identified. 11
SIGAD also may be associated with deficiencies in antipneumococcal antibodies. Drugs. A large number of medications may cause SIGAD, which usually resolves when the medication is discontinued. Examples of these medications include phenytoin, carbamazepine, valproic acid, zonisamide, sulfasalazine, gold compounds, D-penicillamine, hydroxychloroquine, NSAIDs, captopril, and thyroxine.1 In addition, treatment of uveitis with cyclosporin A has been reported to cause permanent IgA deficiency.12
CLINICAL MANIFESTATIONS
Although most persons with SIGAD are asymptomatic, some appear to be more susceptible to particular infections and autoimmune disorders. Up to one-third of patients with SIGAD have recurrent infections (such as recurrent otitis media and sinopulmonary and GI tract infections).1,13 The Table lists the conditions associated with SIGAD.


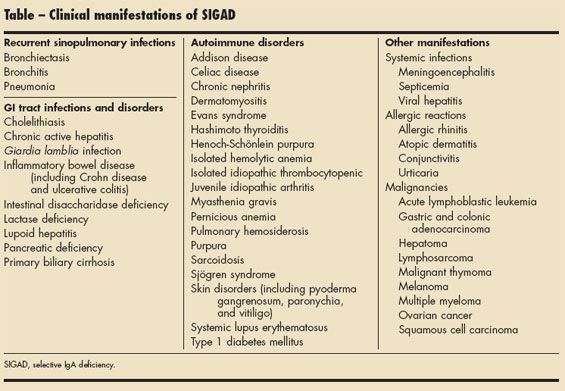
Recurrent sinopulmonary infections. Encapsulated bacteria, including Haemophilus influenzae and Streptococcus pneumoniae, are the most common offending infectious agents. Symptoms may become apparent in the first year of life. Respiratory tract infections may resolve by adulthood in some patients, whereas in other patients, infections may persist throughout adulthood.3 Rarely, recurrent bronchitis, pneumonia, or even bronchiectasis may occur in patients with SIGAD. IgG2 and IgG4 subclass deficiencies are often found in persons who are more severely affected.3
GI tract infections and disorders. In patients with SIGAD, the lack of secretory IgA to bind and destroy toxins and intestinal pathogens leads to a defective mucosal barrier. Consequently, children with SIGAD may have GI tract infections, especially from Giardia lamblia.14 SIGAD is also associated with inflammatory bowel disease, including Crohn disease and ulcerative colitis.15 Other GI diseases, including intestinal disaccharidase deficiency, lactase deficiency, chronic active hepatitis, lupoid hepatitis, primary biliary cirrhosis, cholelithiasis, and pancreatic deficiency, have been reported in association with IgA deficiency.3
Autoimmune disorders. Autoimmune diseases that frequently affect patients with SIGAD include juvenile idiopathic arthritis, systemic lupus erythematosus, dermatomyositis, sarcoidosis, Sjgren syndrome, Evans syndrome, isolated hemolytic anemia, pernicious anemia, isolated idiopathic thrombocytopenic purpura, type 1 diabetes mellitus, Hashimoto thyroiditis, pulmonary hemosiderosis, Addison disease, chronic nephritis, Henoch- Schonlein purpura, and myasthenia gravis.3 Skin disorders, including pyoderma gangrenosum, paronychia, and vitiligo, are also associated with SIGAD.3 The same MHC haplotypes seen in patients with SIGAD and CVID are found in association with some autoimmune disorders, such as type 1 diabetes mellitus and celiac disease.3 It has been theorized that these autoimmune disorders result from recurrent infections with reexposure to foreign antigens that should have been cleared by IgA. Alternatively, the same underlying defect that causes SIGAD may also lead to the development of autoimmune disorders, but the exact pathogenesis remains unclear.3
Other manifestations. Systemic infections, such as viral hepatitis, meningoencephalitis, and septicemia, may also occur in children with SIGAD.3 About 20% of patients with SIGAD have allergic rhinitis, conjunctivitis, urticaria, and atopic dermatitis.3 These may be caused by an increase in IgE to compensate for the IgA deficiency. This elevated IgE level is believed to initiate an allergic or asthmatic component to respiratory problems in persons with SIGAD.
Patients with SIGAD are also predisposed to the development of malignancies, including gastric and colonic adenocarcinoma and acute lymphoblastic leukemia. Other tumors reported in association with SIGAD include multiple myeloma, malignant thymoma, hepatoma, ovarian cancer, lymphosarcoma, squamous cell carcinoma, and melanoma.3
EVALUATION
Recurrent upper and lower respiratory tract infections from encapsulated bacteria in children who are older than 6 months should prompt an evaluation for possible IgA deficiency. This evaluation includes a complete blood cell count and measurement of peripheral blood B cells, T cells, NK cells, and levels of serum immunoglobulins.
Diagnostic criteria. The diagnosis of SIGAD depends on the serum level of IgA, which is compared with that of age-matched controls. Laboratories routinely describe the lowest level of serum IgA as less than 7 mg/dL, because determination of serum levels below this by nephelometry is unreliable.
Partial IgA deficiency is defined as having a serum IgA level more than 2 standard deviations below the mean for age but above 7 mg/dL.3 Low serum IgA levels in a child younger than 4 years may be the result of a transient form of IgA deficiency. After 4 years of age, however, low or absent serum IgA levels are usually considered to be the result of true IgA deficiency.1
Patients with SIGAD have normal serum levels of IgG, IgM, and IgE and normal cell-mediated immunity. A small number of patients with SIGAD may have other abnormalities of immune function, such as deficient production of anticarbohydrate antibodies in response to vaccines or infections.3
SIGAD associated with CVID. Because SIGAD has been found in association with CVID, IgG subclass deficiencies, and impairment of specific antibody responses, the evaluation should not stop with the finding of isolated IgA deficiency. IgG subclass levels should be obtained in patients older than 1 year.16 The measurement of IgG antibodies against gliadin and tissue transglutaminase can also help screen for celiac disease in patients with SIGAD.17,18 Further evaluation of specific antibody responses to polysaccharide and protein antigens should be done to help determine whether these patients would benefit from treatment with intravenous immunoglobulin.9
MANAGEMENT
Patients with SIGAD who have significant recurrent upper respiratory tract infections may benefit from a 6-month course of prophylactic antibiotics. These antibiotics may be continued if the initial course is successful.1 Some children with SIGAD who have seasonal patterns of infections may benefit from receiving prophylactic antibiotics only during the winter months.1 A small number of patients with SIGAD continue to have recurrent infections despite antibiotic prophylaxis. These patients may be treated with immunoglobulin replacement therapy1; however, use of this treatment remains controversial.
IgA Anaphylactic Transfusion Reactions
• A small number of patients with selective IgA deficiency produce IgG or IgE against IgA antibodies. Affected persons are susceptible to adverse transfusion reactions from blood or blood products containing IgA. The estimated incidence of these reactions is about 1 in 20,000 to 47,000 transfusions.19
• Patients who have high anti-IgA levels (more than 1:1000) are predisposed to severe anaphylaxis because they normally have potent antibodies to IgA.3
• Multiparous women or persons who have received multiple transfusions often have low anti-IgA antibody titers (less than 1:256) and may experience minor reactions, including hives and rashes, but severe anaphylaxis rarely develops.
• The levels of anti-IgA antibodies do not always correspond with reactions.19 Exogenous exposures to IgA may not always result in production of anti-IgA antibodies and, in some patien
All persons with SIGAD need to be aware of the risk of severe transfusion reactions resulting from antibodies against IgA (Box)19,20; wearing a medical bracelet is recommended.3 If transfusion is required, other patients with SIGAD are the best donors. Washed, packed red blood cells are considered safer than whole blood transfusion in patients with SIGAD. Monitor the administration of blood products closely so that reactions can be treated promptly.3 Aggressive management of coexisting allergic diseases in children with SIGAD is recommended because allergic inflammation can predispose patients to respiratory tract infections. All children with SIGAD should be immunized according to the same schedule used for children without SIGAD and should receive the annual influenza vaccine. Most patients with SIGAD have an uncomplicated clinical course. However, continued long-term follow-up to identify other immunological abnormalities, such as CVID, autoimmune disease, and malignancy, is required.
References:
REFERENCES:
1. Bonilla FA, Bernstein IL, Khan DA, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. Ann Allergy Asthma Immunol. 2005;94:S1-S63.
2. Boyle JM, Buckley RH. Population prevalence of diagnosed primary immunodeficiency diseases in the United States. J Clin Immunol. 2007;27:497-502.
3. Schroeder HW Jr. Primary antibody deficiencies. In: Rich RR, Fleisher TA, Shearer WT, et al, eds. Clinical Immunology: Principles and Practice. Philadelphia: Mosby; 2008:513.
4. Immunoglobulins and immunoglobulin gene. In: Parslow TG, Stites DP, Terr AI, Imboden JB, eds. Medical Immunology. 10th ed. New York: Lange Medical Books/McGraw-Hill; 2006:102.
5. Adaptive immunity to infection. In: Janeway CA, Travers P, Walport M, Shlomchik MJ, eds. Immunobiology: The Immune System in Health and Disease. 6th ed. New York: Garland Science; 2005:438-439.
6. The humeral immune response. In: Janeway CA, Travers P, Walport M, Shlomchik MJ, eds. Immunobiology: The Immune System in Health and Disease. 6th ed. New York: Garland Science; 2005:390.
7. Schaffer FM. Clinical assessment and management of abnormal IgA levels. Ann Allergy Asthma Immunol. 2008;100:280-282.
8. Rachid R, Castigli E, Geha RS, Bonilla FA. TACI mutation in common variable immunodeficiency and IgA deficiency. Cur Allergy and Asthma Rep. 2006;6:357-362.
9. Hammarström L, Vorechovsky I, Webster D. Selective IgA deficiency (SIgAD) and common variable immunodeficiency (CVID). Clin Exp Immunol. 2000;120:225-231.
10. Vorechovsky I, Webster AD, Plebani A, Hammarström L. Genetic linkage of IgA deficiency to the major histocompatibility complex: evidence for allele segregation distortion, parent-of-origin penetrance differences, and the role of anti-IgA antibodies in disease predisposition. Am J Hum Genet. 1999;64:1096–1109.
11. Johnson ML, Keeton LG, Zhu ZB, et al. Age-related changes in serum immunoglobulins in patients with familial IgA deficiency and common variable immunodeficiency (CVID). Clin Exp Immunol. 1997;108:477-483.
12. Murphy EA, Morris AJ, Walker E, et al. Cyclosporine A induced colitis and acquired selective IgA deficiency in a patient with juvenile chronic arthritis. J Rheumatol. 1993;20:1397-1398.
13. Cunningham-Rundles C. Physiology of IgA and IgA deficiency. J Clin Immunol. 2001;21:303-309.
14. Zinneman HH, Kaplan AP. The association of giardiasis with reduced intestinal secretory immunoglobulin A. Am J Dig Dis. 1972;17:793-797.
15. Manfredi R, Coronado OV, Marinacci G, et al. Crohn’s disease, rare association with selective IgA immunodeficiency, and development of life-threatening bacterial infections. Scand J Infect Dis. 2004;36:523-524.
16. Sandler SG, Trimble J, Mallory DM. Coexisting IgG2 and IgA deficiencies in blood donors. Transfusion. 1996;36:256-258.
17. Cataldo F, Marino V, Bottaro G, et al. Celiac disease and selective immunoglobulin A deficiency. J Pediatr. 1997;131:306-308.
18. Deiterich W, Laag E, Schöpper H, et al. Autoantibodies to tissue transglutaminase as predictors of celiac disease. Gastroenterology. 1998;115:1317-1321.
19. Sandler SG, Mallory D, Malamut D, Eckrich R. IgA anaphylactic transfusion reactions. Transfus Med Rev. 1995;9:1-8.
20. Ferreira A, Garcia Rodriguez MC, Lopez- Trascasa M, et al. Anti-IgA antibodies in selective IgA deficiency and in primary immunodeficient patients treated with gamma-globulin. Clin Immunol Immunopathol. 1988;47:199-207.



















