



New-Onset Seizures in Infant With Square Facies, Hypospadias, and Hirschsprung Disease
A 6-month-old boy with dysmorphic facial features, ear anomalies, and hypospadias brought for evaluation of elevated temperature and generalized seizure. Infant born at term to a 37-year-old mother via uncomplicated cesarean delivery.


HISTORY
A 6-month-old boy with dysmorphic facial features, ear anomalies, and hypospadias brought for evaluation of elevated temperature and generalized seizure. Infant born at term to a 37-year-old mother via uncomplicated cesarean delivery. In the nursery, progressive abdominal distention and bilious vomiting developed. Barium enema revealed findings consistent with Hirschsprung disease. Infant underwent corrective surgery with colostomy and subsequent colostomy reversal. A moderate ventricular septal defect was detected during his neonatal course and confirmed by echocardiogram. CT scan of the head revealed agenesis of the corpus callosum.
PHYSICAL EXAMINATION
Square-shaped face, a prominent but narrow chin, deep-set but large eyes, hypertelorism, broad nasal bridge with rounded nasal tip, open mouth with full lower lip, posteriorly rotated ears, and large uplifted earlobes with a central depression. Infant's weight and height at the 25th percentile; head circumference at the 50th percentile. Large horizontal scar from surgery on his abdomen, and a harsh grade 3/6 systolic ejection murmur over left sternal area. Vital signs normal.
WHAT'S YOUR DIAGNOSIS?
Answer on Next Page
ANSWER: MOWAT-WILSON SYNDROME
Mowat-Wilson syndrome (MWS) is a relatively new intellectual disability syndrome characterized by unique facial features and a common association with Hirschsprung disease. It was first described in 1998 by Mowat and colleagues,1 who found several cytogenic deletions in chromosome 2 at 2q21-23 in 6 patients with similar clinical features. Overall, the cause of the syndrome is by de novo heterozygous mutations or deletions of the ZFHX1B gene, now known as ZEB2, or zinc finger E-box-binding-homeobox 2 gene.2
INCIDENCE
The incidence of MWS is currently unknown; however, the syndrome is likely underdiagnosed, especially in patients without the classic Hirschsprung findings. Since the first report in 1998, ZEB2 mutations, deletions, or cytogenetic abnormalities have been reported in 171 patients primarily from Northern Europe,


Australia, Italy, and the United States; more than 100 mutations have been described.3 The male to female ratio is about 1.4:1. The syndrome has been identified in several ethnic groups, with similar clinical features in all populations.4
CLINICAL FEATURES
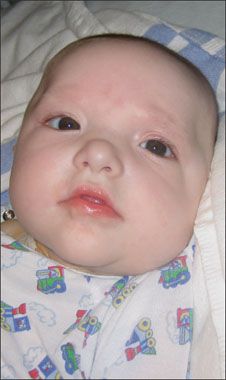
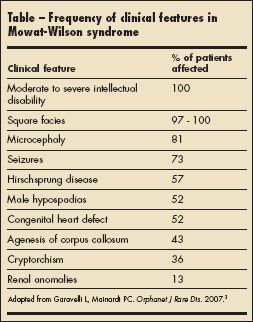
Most patients with MWS have moderate to severe intellectual disability with absent or severely impaired speech. Gross and fine motor milestones are generally delayed. A square-shaped face with a small triangular chin is the most common facial anomaly (Table). Other facial features include orbital hypertelorism, which is associated with deeply set eyes, a broad nasal bridge with prominent rounded nasal tip, and a full or everted lower lip. The ears may be posteriorly rotated, with large uplifted earlobes and a central depression (Figure 1). Common congenital anomalies include Hirschsprung disease, heart defects, hypospadias (Figure 2), genitourinary abnormalities, microcephaly, agenesis of the corpus callosum, and short stature.1-4 Seizures develop in about 75% of affected patients, usually by 2 years of age.5


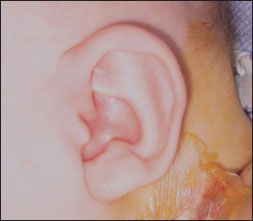
Figure 1 – Infants with Mowat-Wilson syndrome may have posteriorly rotated ears and large uplifted earlobes with a central depression.
DIAGNOSIS
Because of its early presentation, MWS is usually diagnosed in infants with Hirschsprung disease and the typical facial phenotype. However, Hirschsprung disease is not present in all infants with this syndrome and thus is not required for diagnosis. The characteristic facial phenotype of MWS differentiates it from other genetic syndromes, such as Goldberg-Shprintzen syndrome, Angelman syndrome, and Smith-Lemi-Opitz syndrome. Documentation of ZEB2 gene mutations with genetic testing is not required for diagnosis; however, it is becoming more available and should be done.3 Confirming the diagnosis may be helpful in family planning and can alert the physician and parents to be aware of associated findings.
GENETICS
The ZEB2 gene encodes for Smad interacting protein-1 (SIP1), which is a transcription factor involved in the transforming growth factor β signaling pathway.4 SIP1 is thought to play a crucial role in embryonic development. Animal studies on the homologues of SIP1 show that SIP1 expression is active in early embryogenesis, including neural tube and neural crest development.
MANAGEMENT
Because of the many congenital malformations associated with MWS, a clinical evaluation with multiple subspecialists is required. Hirschsprung disease usually requires early surgery in the first days or months of life with aggressive management for constipation. Congenital heart disease needs to be evaluated for severity and may require surgery or close follow-up with a cardiologist. Seizures can begin within the first few months of life and may require antiseizure medications for a prolonged period. Genitourinary anomalies, such as hypospadias, cryptorchism, bifid scrotum, vesicoureteral reflux, and hydronephrosis, may be present in the first years of life and may also require surgery. Early intervention with speech and physical therapy is required for all children with MWS.


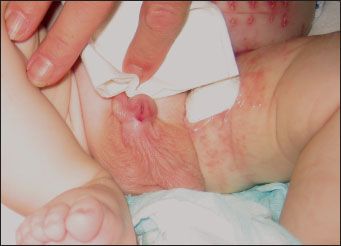
Figure 2 – Hypospadias is a common congenital anomaly associated with Mowat-Wilson syndrome.
PROGNOSIS
Few data are available on the prognosis of MWS. Confirmation of the diagnosis with molecular testing is important, because the earlier rehabilitation services are started, the better. Although most cases of MWS are sporadic, some sibling recurrence has been observed.3 This patient's parents were linked with an international MWS support group provided by our geneticist.
References:
REFERENCES:1. Mowat DR, Croaker GD, Cass DT, et al. Hirschsprung disease, microcephaly, mental retardation, and characteristic facial features: delineation of a new syndrome and identification of a locus at chromosome 2q22-q23. J Med Genet. 1998;35:617-623.
2. Cerruti Mainardi P, Pastore G, Zweier C, Rauch A. Mowat-Wilson syndrome and mutation in the zinc finger homeo box 1B gene: a well defined clinical entity. J Med Genet. 2004;41:e16.
3. Garavelli L, Mainardi PC. Mowat-Wilson syndrome. Orphanet J Rare Dis. 2007;2:42.
4. Dastot-Le Moal F, Wilson M, Mowat D, et al. ZFHX1B mutations in patients with Mowat-Wilson syndrome. Hum Mutat. 2007;28:313-321.
5. Adam MP, Schelley S, Gallagher R, et al. Clinical features and management issues in Mowat-Wilson syndrome. Am J Med Genet A. 2006;140:2730-2741.








Seizures could be the cause of many sudden unexplained deaths in toddlers
January 9th 2024The study authors noted that without video evidence, seizures would not have been implicated in death investigations and that seizure-related deaths are underrecognized in patients with epilepsy and in those without.

FDA warns of serious potential reaction to levetiracetam and clobazam
November 30th 2023The reaction, called Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS), can start as a rash but can progress quickly, potentially resulting in injury to internal organs, hospitalization, and death.

Dietary therapies safe and efficient for treating drug-resistant epilepsy
February 7th 2023While treatment discontinuation because of adverse events was more likely when taking dietary therapies for treating childhood drug-resistant epilepsy, reduction of seizures and seizure freedom were significantly more common.



