



Genetic Disorders: Child With Facial Anomalies and Developmental Delay
Genetic Disorders: Child With Facial Anomalies and Developmental Delay



An 18-month-old girl with growth and developmental delay was referred for geneticevaluation. The child had had an unremarkable term gestation but a low birth weight of 5 lb (2.25 kg). She had difficulty in latching on for breast-feeding but had no other problems.
By the time the child was 6 months old, her pediatrician was concerned about her slow motor development and poor head control. At 2 months, she was able to roll over front-to-back; at 4 months, she could roll from back to front but was not able to maintain the sitting position. At 1 year, she was making progress but development was still delayed; pediatric and neurology evaluations documented central hypotonia. An MRI scan of the head demonstrated delayed myelination. The patient had a normal thyroid and had undergone testing for cystic fibrosis.
Physical examination at age 14 months revealed that the child's length, weight, and head circumference were proportionate but were below the third percentile for age and average for a 2-month-old. The child's face was remarkable for mild ptosis, wide palpebral fissures, epicanthal folds, large ears with over-folded helices, and an open mouth with down-turned corners. Her palate revealed broad alveolar ridges and her finger pads were prominent. She babbled but said no words. Her estimated developmental level was 6 to 8 months.
TO WHAT DIAGNOSIS DO THESE CLINICAL FINDINGS POINT?


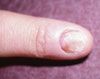
Figure 1


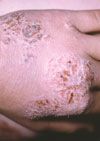
Figure 2
ANSWER: KABUKI SYNDROME
In 1981, 2 Japanese groups reported a characteristic malformation pattern with growth and developmental delay.1,2 Major findings included an unusual facial appearance that was likened to a Kabuki theater mask (Figures A and B); prenatal and postnatal growth delay with short stature; mild to moderate mental retardation; dermatoglyphic abnormalities with frequent ulnar loops and prominent "fetal" finger pads (Figure C); and skeletal anomalies, including scoliosis, vertebral anomalies, and congenital hip dislocation. The Niikawa-Kuroki or Kabuki syndrome became well-appreciated in Japan, with an incidence of about 1 in 32,000 births. Reported cases in occidental populations became common in the mid-1990s: reports emphasized joint laxity, cardiac lesions, and urogenital anomalies as additional components of the Kabuki spectrum.3
There are now more than 250 reported cases of Kabuki syndrome.4 This suggests that its incidence in the West (1 in 10,000 to 20,000 births) compares with that of other syndromes the pediatrician should recognize (ie, the Williams or Prader-Willi syndromes). Occidental patients do not resemble a Kabuki theater mask, but they may have an Asian appearance with large eyes, large ears, and open mouth. The delayed growth and development and hypotonia (open mouth, joint laxity, broad alveolar ridges from decreased fetal tongue movement), together with the large eyes and ears, produce a distinctive clinical picture.
Figure 1
A comprehensive recent review4 emphasizes that there is still no definitive laboratory test for Kabuki syndrome because its cause remains unknown. The equal sex ratio, sporadic nature of most cases, and occasional instances of vertical transmission suggest autosomal dominant inheritance with frequent de novomutation. The diagnosis is clinical: 5 cardinal manifestations serve the clinician until consensus criteria are defined.1,2,4 The characteristic face is paramount, with arched eyebrows (in 85% of those affected), long palpebral fissures (99%), prominent ears (84%), short nasal septum (92%), and depressed nasal tip (83%).
The other 4 manifestations are skeletal anomalies, such as short or curved fifth finger (clinodactyly in 79%) and scoliosis (35%); dermatoglyphic changes, including prominent fingertip pads (89%); mild to moderate mental disability (80% have an IQ score under 80); and growth deficiency (55%).3 Recognition is possible in the newborn period.5
Several cases of Kabuki syndrome have been associated with chromosome aberrations--including inverted or ring Y, rearrangements of chromosomes 4, 13, or 15/17 and, most consistently, X chromosome rearrangements, including ring X, 45,X Turner syndrome, and low-grade 45,X mosaicism. The chromosome 22q11 deletion that causes the DiGeorge-Shprintzen spectrum has been excluded in several patients. One report used the new techniques of comparative genomic hybridization (CGH--screening the patient's DNA for extra or missing genes) and fluorescent in situ hybridization (FISH--screening the patient's chromosomes for small extra [3 signals] or missing fragments [1 signal]) to suggest a microduplication of chromosome 8 in Kabuki syndrome.6 However, numerous recent reports have not substantiated this finding. Nevertheless, it may be useful to examine suspect cases of Kabuki; newer cytogenetic analyses, such as subtelomere FISH, help exclude similar phenotypes caused by subtle chromosome rearrangements.
Recognition of Kabuki syndrome confers several advantages to the patient and family. These include an explanation of the growth/developmental delay and a clearer understanding of the low recurrence risks in future pregnancies. Anticipatory guidance can be supplemented with knowledge of many potential complications, including craniosynostosis, strabismus, palatal clefts or velopalatine incompetence, chronic otitis with hearing deficits, hypothyroidism, cardiac septal defects, mitral valve prolapse or insufficiency, aortic coarctation, immune and immunoglobulin deficiencies, pectus excavatum, gastroesophageal reflux, deep sacral dimple with tethered cord, hemivertebrae, imperforate anus, congenital dislocated hips, micropenis or cryptorchidism, inguinal hernia, horseshoe kidneys, hydronephrosis, renal duplication, seizures, and brain anomalies (including delayed myelination or absent septum pellucidum, absent corpus callosum, and arachnoid cysts).3,4
Intelligence level may be nearly normal to severely impaired (often foretold by disproportionate microcephaly). Behavioral problems include short attention span, hyperactivity, and autistic spectrum disorder. Neonatal hypoglycemia is frequent: several mothers of children with Kabuki syndrome have had diabetes mellitus.
MANAGEMENT
Preventive management should include early monitoring of feeding because of frequent palatal problems and dysphagia from hypotonia. Referral for early intervention and, in particular, for speech therapy is mandated because of early oromotor problems and later dysarthria, dyspraxia, and hypernasal speech.
Lax connective tissue should be anticipated, as evidenced by blue sclerae, joint laxity, patellar dislocation, pectus excavatum, hip dislocation, or even aortic aneurysm. Alertness for skeletal anomalies is important because cervical vertebral fusions or thoracic anomalies (such as elevated diaphragm or diaphragmatic hernia) may complicate surgeries.
Although growth delay can be expected, patients with severe failure to thrive may warrant a gastroenterology evaluation for severe reflux, malrotation, or rectovaginal fistula.
Renal imaging for urogenital anomalies should be considered along with endocrinology referral for neonatal hypoglycemia/hypocalcemia and elevated follicle-stimulating hormone (with premature thelarche), deficient growth hormone, or diabetes insipidus.
Cardiac anomalies, such as septal defects or aortic coarctation/bicuspid aortic valve, are common enough that an infantile echocardiogram can be justified--regardless of symptoms. Hearing and vision must be monitored carefully because of the risk of strabismus or conductive hearing deficits (from chronic respiratory infections augmented by immune deficiency).
References:
REFERENCES:
1.
Niikawa N, Matsuura N, Fukushima Y, et al. Kabuki make-up syndrome: a syndrome of mental retardation, unusual facies, large and protruding ears, and growth deficiency.
J Pediatr.
1981;99:565-569.
2.
Kuroki Y, Suzuki Y, Chyo H, et al. A new malformation syndrome of long palpebral fissures, large ears, depressed nasal tip, and skeletal anomalies associated with postnatal dwarfism and mental retardation.
J Pediatr.
1981;99:570-573.
3.
Wilson GN. Thirteen cases of Niikawa-Kuroki syndrome: report and review with emphasis on medical complications and preventive management.
Am J Med Genet.
1998;79:112-120.
4.
Adam MP, Hudgins L. Kabuki syndrome: a review.
Clin Genet.
2005;67:209-219.
5.
Vaux KK, Hudgins L, Bird LM, et al. Neonatal phenotype in Kabuki syndrome.
Am J Med Genet A.
2005;132:244-247.
6.
Milunsky JM, Huang XL. Unmasking Kabuki syndrome: chromosome 8p22-8p23.1 duplication revealed by comparative genomic hybridization and BAC-FISH.
Clin Genet.
2003;64:509-516.


















